DETERMINATION OF VOLUME OF INJECTION IN CONTAINERS
Select 1 or more containers if the volume of the container is 10 mL or more, 3 or more if the volume is more than 3 mL and less than 10 mL, or 5 or more if the volume is 3 mL or less. Individually take up the contents of each container selected into a dry hypodermic syringe of a rated capacity not exceeding three times the volume to be measured and fitted with a 21-gauge needle not less than 2.5 cm (1 inch) in length. Expel any air bubbles from the syringe and needle, and then discharge the contents of the syringe, without emptying the needle, into a standardized, dry cylinder (graduated to contain rather than to deliver the designated volumes) of such size that the volume to be measured occupies at least 40% of the cylinder's rated volume. Alternatively, the contents of the syringe may be discharged into a dry, tared beaker, the volume, in mL, being calculated as the weight, in g, of Injection taken divided by its density. For containers with a nominal volume of 2 mL or less, the contents of a sufficient number of containers may be pooled to obtain the volume required for the measurement, provided that a separate, dry syringe assembly is used for each container. The content of containers holding 10 mL or more may be determined by means of opening them and emptying the contents directly into the graduated cylinder or tared beaker.
The volume is not less than the labeled volume in the case of containers examined individually or, in the case of 1- and 2-mL containers, is not less than the sum of the labeled volumes of the containers taken collectively.
For Injections in multiple-dose containers labeled to yield a specific number of doses of a stated volume, proceed as directed in the foregoing, using the same number of separate syringes as the number of doses specified. The volume is such that each syringe delivers not less than the stated dose.
For Injections containing oil, warm the containers, if necessary, and thoroughly shake them immediately before removing the contents. Cool to between 20 and 25
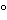
before measuring the volume.
For Injections in cartridges or prefilled syringes, assemble the container with any required accessories such as a needle or plunger. Following the same procedure as above, and without emptying the needle, transfer the entire contents of each container to a dry, tared beaker by slowly and constantly depressing the plunger. Weigh, and calculate the volume as described above. The volume of each container is not less than the labeled volume.
For large-volume intravenous solutions, select 1 container, and transfer the contents into a dry measuring cylinder of such size that the volume to be measured occupies at least 40% of its rated volume. The volume is not less than the labeled volume.